
守正创新 终结艾滋 | HIV-1 全基因组检测一体化解决方案
01 HIV-1 全基因组检测为何成为艾滋病防控核心?
人类免疫缺陷病毒 (Human Immunodeficiency Virus,HIV) 是引发全球性重大恶性传染病 —— 艾滋病 / 获得性免疫缺陷综合征 (Acquired Immunodeficiency Syndrome,AIDS) 的主要病原体,至今仍是全球公共卫生防控面临的重大挑战。造成这一困境的核心原因在于 HIV 极高的遗传变异性和快速进化能力。HIV 以 RNA 为遗传物质,在复制过程中依赖缺乏校对功能的逆转录酶,变异率极高;同时,病毒在宿主体内高速复制并频繁发生基因重组,在免疫压力和药物选择压力的共同作用下,迅速形成高度多样化的准种群。这种持续演化的特性,使得 HIV 如同一个不断“变身”的狡猾敌人,不仅能够逃避免疫系统的监视,还能在治疗过程中快速产生耐药性,导致疗效下降甚至治疗失败,以及耐药毒株传播,给临床管理和群体防控带来长期挑战。
面对越来越狡猾的 HIV,该如何应对?
根据基因差异,HIV 可分为 HIV-1 型和 HIV-2 型。其中,HIV-1 型是全球艾滋病流行的主要毒株,传播力和致病性更强,遗传多样性也最为复杂;HIV-2 型主要局限于非洲西部及部分欧洲地区,传播效率和致病性相对较低。其中,在 HIV-1 型内还存在至少 10 种基因亚型和流行重组型,我国 HIV-1 流行最广泛的毒株为 CRF01_AE,CRF07_BC 重组型,以及 B’ 亚型,但不同地区基因亚型存在差异,其分布与流行特征直接影响诊断、治疗策略和防控措施的制定。
在此背景下,针对 HIV-1 型病毒的全基因组检测则显得尤为重要,其能够在艾滋病防控中发挥出不可替代的核心价值,对精准鉴定病毒亚型与重组型、快速追踪传播链、支持临床诊断决策、监测抗病毒治疗效果以及推动新型诊断试剂、抗病毒药物和疫苗研发等方面均具有重要意义。
相较于传统核酸检测方法,基于纳昂达独家专利技术的 μCaler® HIV-1 全基因组检测一体化解决方案展现出显著优势。该方案可完整覆盖 HIV-1 约 9.7 Kb 的全长基因组,避免因重复序列或重组事件导致的拼接或组装错误,尤其在变异监测、精准分型、准种鉴定、感染溯源和耐药分析等方面优势突出,为 HIV 感染病例的精准防控和长期规范化管理提供了有力的技术支撑。
02 HIV-1 全基因组检测方案
2.1 方案简介
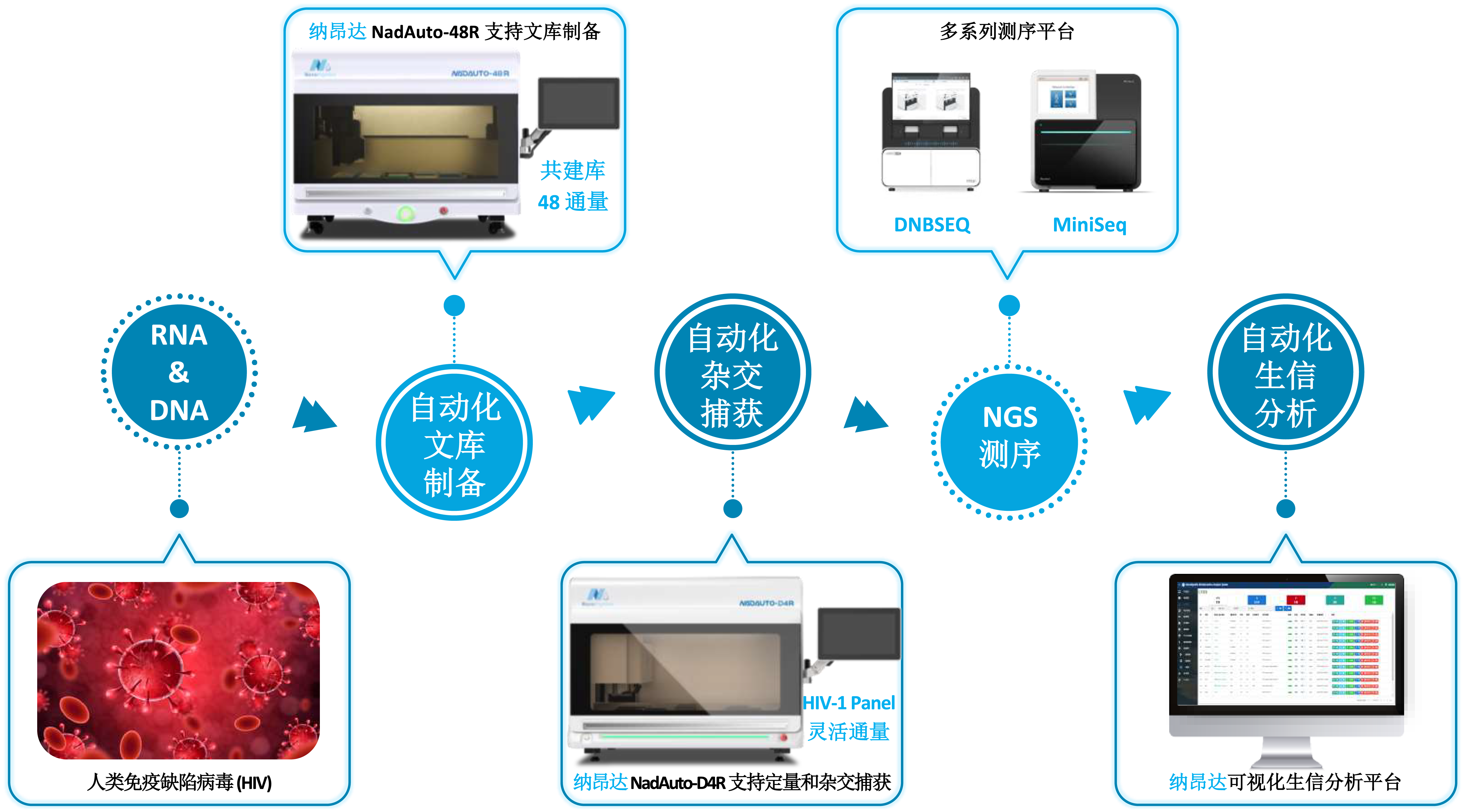
μCaler® HIV-1 全基因组检测一体化解决方案基于快速 RNA & DNA 共建库试剂盒,结合μCaler® 杂交捕获系统与 μCaler® HIV-1 Panel v1.0,可高效构建高质量 NGS 文库,并兼容主流测序平台开展测序分析。测序数据下机后,配合 XCapViz 生信分析可视化系统及在线分析网站,可在单日内实现“样本进,结果出”,为 HIV-1 全基因组序列的正确拼接与组装、分型鉴定及变异动态追踪提供全方位技术支持,有效助力 HIV 感染病例的快速精准诊疗与流行病学的监测管理。
同时,依托纳昂达灵活多样的 NadAuto 系列全自动 NGS 工作站也可进一步简化实验操作流程,降低人工干预强度,释放实验室人力资源,提升临床检测通量与效率,并减少人为误差及职业暴露风险,为 HIV 流行病学溯源研究和传播风险评估提供高效、可靠的方案支持。
2.2 特色亮点
● 极速检测:3 hr 快速建库 (支持 RNA 单核酸样本投入),1 hr 极速杂交,显著提升 NGS 文库制备的效率;
● 全长覆盖:基于全球流行的优势 HIV-1 毒株参考序列设计基因组全长探针,支持完整解析全长基因组序列;
● 精确解析:全基因组分析较单 / 多基因分型更易识别复杂的流行重组事件并提供更全面的耐药位点注释信息。
03 HIV-1 全面解析 精准分型
3.1 完整覆盖全基因组
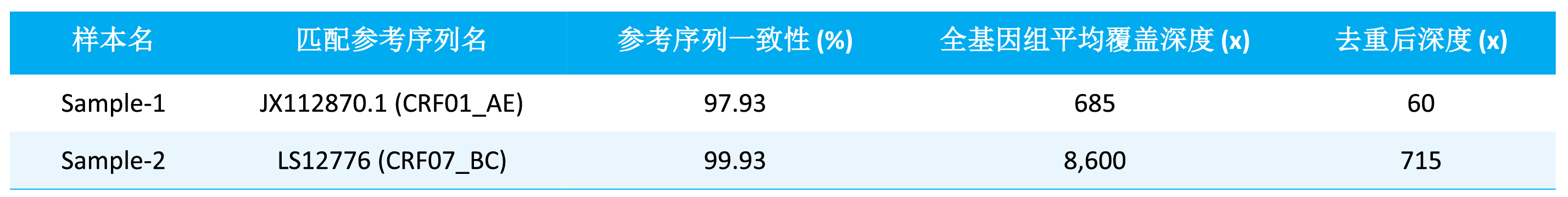
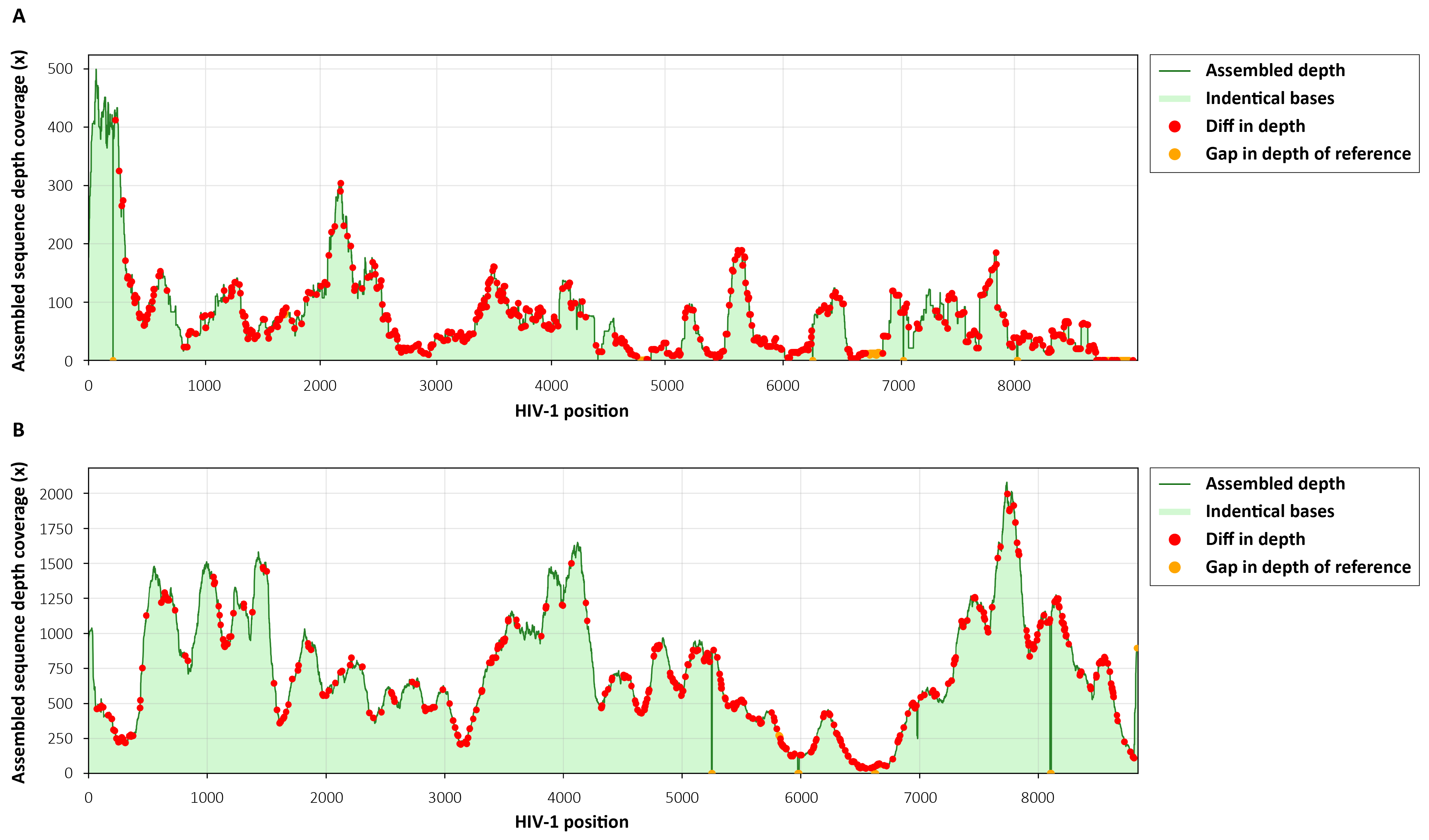
为了评估 μCaler® HIV-1 全基因组检测一体化解决方案在 HIV-1 全长基因组拼接与组装方面的表现,我们对测序组装后的全长基因组序列与参考序列的一致性及全基因组覆盖深度进行了分析。将 10 ng Carrier RNA (Cat # 1005601) 与 2 例临床来源的 HIV 核酸样本混合,利用快速 RNA & DNA 文库构建试剂盒构建预文库,以 μCaler® Hybrid Capture Reagents v2 和 μCaler® HIV-1 Panel v1.0 完成杂交捕获并上机测序。结果显示,该方案对不同亚型的 HIV-1 均能获得高覆盖度的全长基因组序列,且与参考序列保持高一致性 (表 1. & 图 3.),为后续的深度变异和流行重组型分析、传播链追踪,以及耐药注释提供了可靠的数据基础。
3.2 精准鉴定与分型
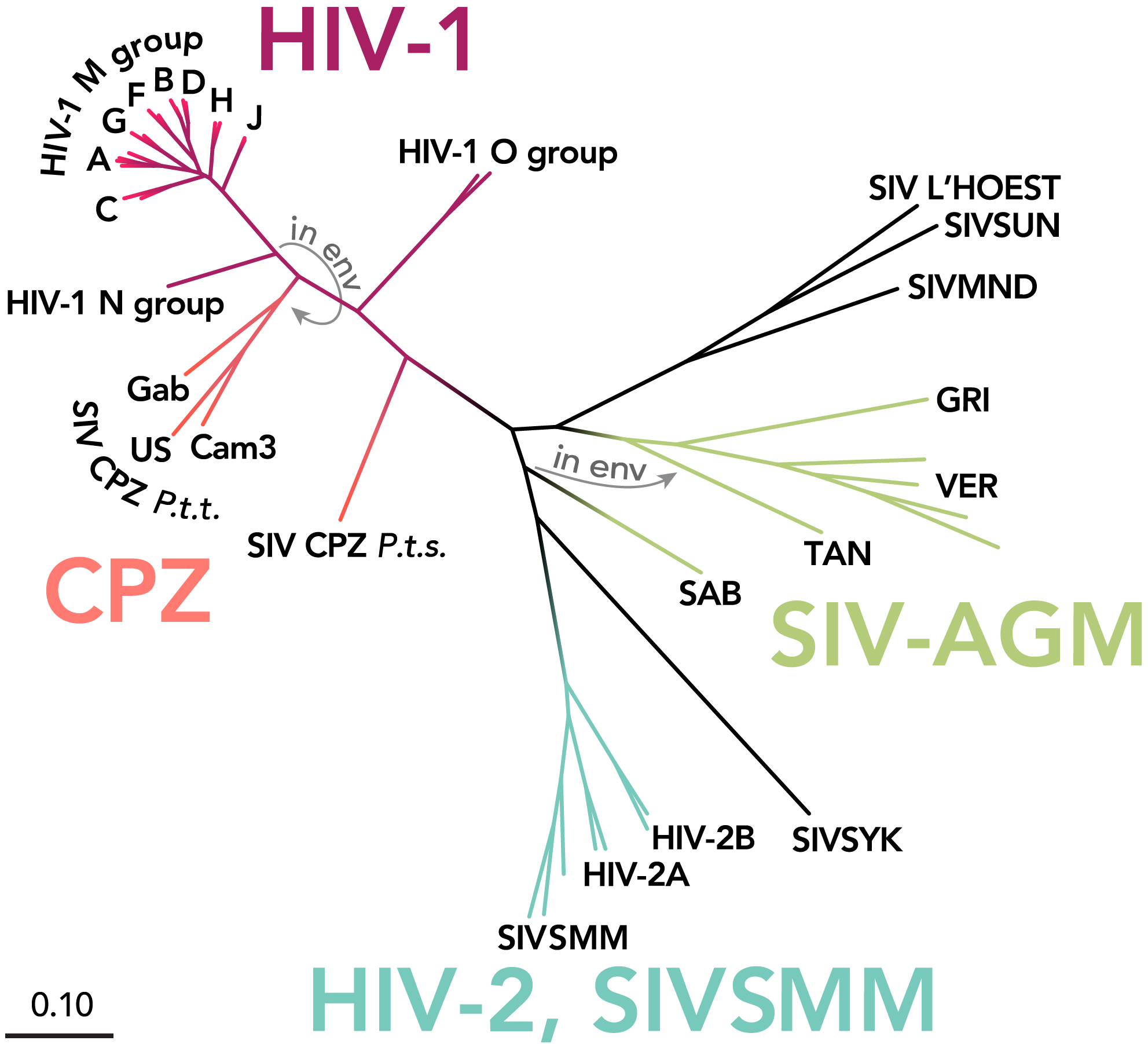
HIV-1 型可依据病毒进化特征分为 4 个亚型组,分别为 M (major)、N (non-M, non-O)、O (outlier) 以及可能存在的 P 组。其中,M 组在全球范围内广泛流行 (约占全球 HIV 感染病例的 90%)。 该组病毒又可进一步细分为 10 种以上的 HIV-1 亚型 (A、B、C、D、F、G、H、J、K 和 L),以及大量流行重组型 (Circulating Recombinant Form,CRF) 和独特重组型 (Unique Recombinant Form,URF),构成了极其复杂的遗传多样性谱系。从基因组结构上看,HIV-1 结构基因由 pol、env、gag 组成,均可用于亚型鉴定。目前,获取亚型信息最为全面的方法是对 HIV-1 全长基因组或部分基因区域 (如 pol、env、gag) 进行序列测定。由于耐药研究和临床监测的广泛开展,pol 区域成为目前最常用的测序与分型分析区域。
μCaler® HIV-1 全基因组检测一体化解决方案结合全长基因组序列与 pol 基因分析,已成功鉴定出多种亚型和流行重组型,包括:B, C, CRF01_AE, CRF07_BC, CRF08_BC, CRF55_01B, CRF59_01B, CRF65_cpx, CRF68_01B 和 CRF85_BC。以 2 例临床来源的 HIV 核酸样本为代表进行亚型分析展示 (表 2.),结果显示,部分样本在基于全长基因组序列的亚型判定结果与仅基于 pol 基因的分型结果之间存在不一致性,这类差异通常提示该病毒株可能为多亚型重组病毒。
为进一步验证上述推断,以 Sample-2 为例开展深入分析。镶嵌图结果显示,该毒株基因组由不同亲本亚型片段交替嵌合而成。Bootscan 分析进一步证实,在不同基因组区段内,该毒株序列与潜在亲本亚型参考序列的 Bootstrap 值 (节点支持值) 出现显著切换,且切换节点与镶嵌图中预测的重组断点高度一致。在多个区段中观察到的较高 Bootstrap 值 (置信区间通常 > 70%),从统计学层面明确了各基因区段的亚型归属 (图 4.)。
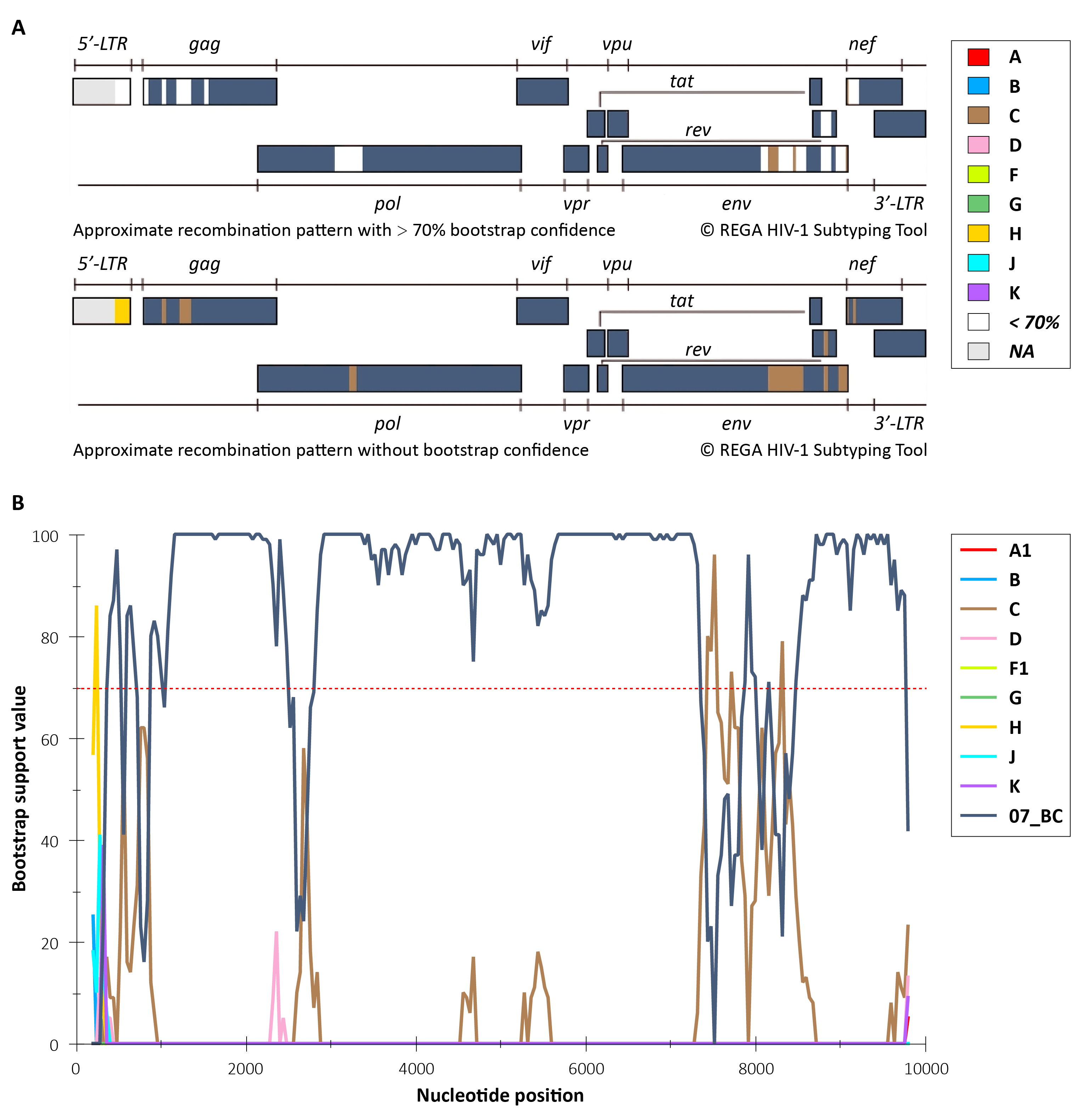
上述结果表明,μCaler® HIV-1 全基因组检测能够更加全面、准确地揭示病毒复杂的重组背景,而仅依赖单一基因片段区域 (如 pol) 的分析,可能难以完整反映此类重组特征。因此,快速且精准的全基因组分型鉴定对 HIV-1 分子流行病学管理、抗逆转录病毒治疗监测以及艾滋病全球防控等均具有重要意义。
04 总结与展望
μCaler® HIV-1 全基因组检测一体化解决方案,基于快速 RNA & DNA 共建库、独家专利技术的 μCaler® 杂交捕获系统及 XCapViz 可视化生信分析体系与在线分析网站,可对复杂且高度多样化的 HIV-1 全长基因组进行高覆盖、系统性解析,精准识别多亚型重组事件,显著提升亚型判定的准确性与分子溯源能力,为传播链追踪、 耐药突变注释及临床决策提供可靠而高效的工具支撑。
展望未来,随着 HIV 分子流行病学监测不断向精细化、系统化方向发展,以及个体化治疗需求的持续增长,μCaler® HIV-1 全基因组检测一体化解决方案有望在疾病监测网络建设、耐药风险评估及科研成果转化等多元应用场景中发挥更大价值,为艾滋病的长期精准防控提供持续、稳健的技术支持,助力实现终结艾滋病流行的全球目标。






